A new antifungal molecule, devised by tweaking the structure of prominent antifungal drug Amphotericin B, has the potential to harness the drug’s power against fungal infections while doing away with its toxicity, researchers at the University of Illinois Urbana-Champaign and collaborators at the University of Wisconsin-Madison report in the journal Nature.
Amphotericin B, a naturally occurring small molecule produced by bacteria, is a drug used as a last resort to treat fungal infections. While AmB excels at killing fungi, it is reserved as a last line of defense because it also is toxic to the human patient – particularly the kidneys.
“Fungal infections are a public health crisis that is only getting worse. And they have the potential, unfortunately, of breaking out and having an exponential impact, kind of like COVID-19 did. So let’s take one of the powerful tools that nature developed to combat fungi and turn it into a powerful ally,” said research leader Dr. Martin D. Burke, an Illinois professor of chemistry, a professor in the Carle Illinois College of Medicine and also a medical doctor.
“This work is a demonstration that, by going deep into the fundamental science, you can take a billion-year head start from nature and turn it into something that hopefully is going to have a big impact on human health,” Burke said.
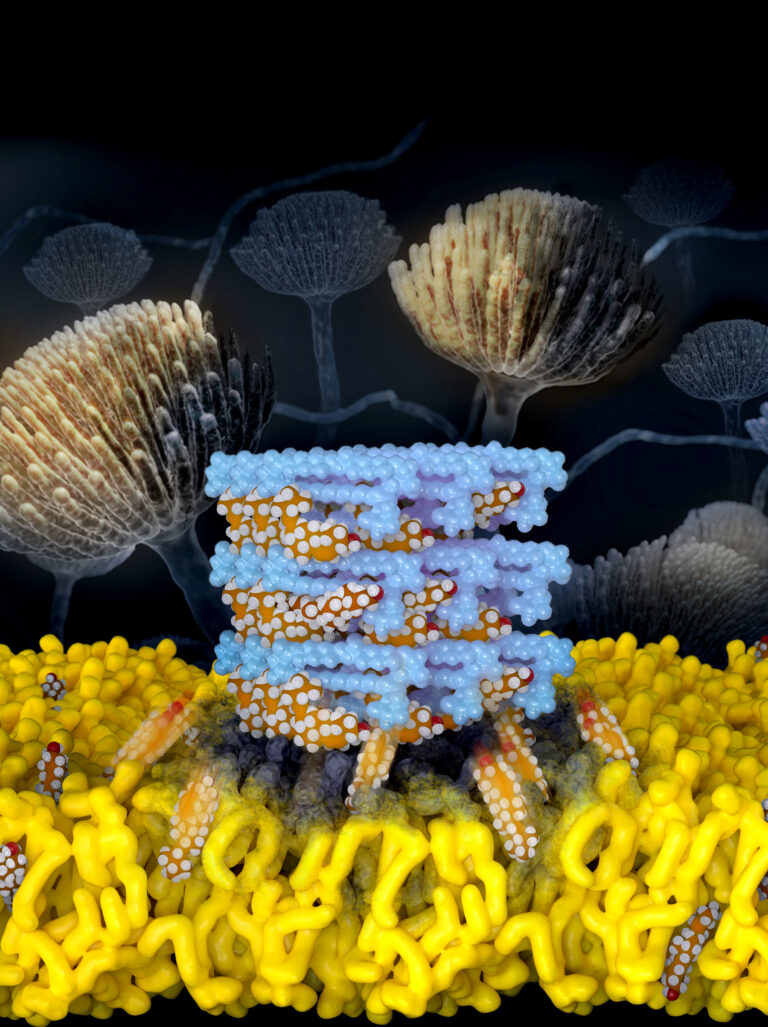
The mechanism for a critical but highly toxic antifungal is revealed in high resolution. Self-assembled Amphotericin B sponges rapidly extract sterols from cells. This atomic level understanding yielded a breakthrough renal sparing polyene antifungal. Image by Jose Vazquez.